Proteins
- Conformation of proteins.
- Mechanical Unfolding of polydomain protein concatamers.
- Monte Carlo and Molecular Dynamics Simulations
- (2002-2011)Funded through Wellcome Trust, in collaboration with DJ Brockwell
(Leeds Biochemistry), SE Radford (Leeds Biochemistry), E
Paci (Leeds Biochemistry), and DA Smith (Leeds Physics).
- (2014) Funded from the Swiss National Science Foundation, in Collaboration
with R Mezzenga (ETH Zurich).
Papers:
Amyloid fibril bending and ring formation at liquid
interfaces
Sophia Jordens, Emily E. Riley, Ivan Usov, Lucio
Isa, Peter D. Olmsted, and Raffaele Mezzenga,
ACS Nano 8 (2014)
11071-11079.
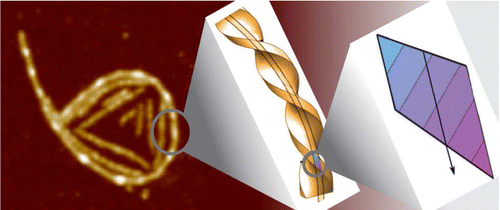 The Protein fibril accumulation at interfaces is an
important step in many physiological processes and neurodegenerative
diseases as well as in designing materials. Here we show, using
beta-lactoglobulin fibrils as a model, that semiflexible fibrils exposed
to a surface do not possess the Gaussian distribution of curvatures
characteristic for wormlike chains, but instead exhibit a spontaneous
curvature, which can even lead to ring-like conformations. The
long-lived presence of such rings is confirmed by atomic force
microscopy, cryogenic scanning electron microscopy, and passive probe
particle tracking at air! and oil!water interfaces. We reason that
this spontaneous curvature is governed by structural characteristics
on the molecular level and is to be expected when a chiral and polar
fibril is placed in an inhomogeneous environment such as an
interface. By testing β-lactoglobulin fibrils with varying average
thicknesses, we conclude that fibril thickness plays a determining
role in the propensity to form rings.
The Protein fibril accumulation at interfaces is an
important step in many physiological processes and neurodegenerative
diseases as well as in designing materials. Here we show, using
beta-lactoglobulin fibrils as a model, that semiflexible fibrils exposed
to a surface do not possess the Gaussian distribution of curvatures
characteristic for wormlike chains, but instead exhibit a spontaneous
curvature, which can even lead to ring-like conformations. The
long-lived presence of such rings is confirmed by atomic force
microscopy, cryogenic scanning electron microscopy, and passive probe
particle tracking at air! and oil!water interfaces. We reason that
this spontaneous curvature is governed by structural characteristics
on the molecular level and is to be expected when a chiral and polar
fibril is placed in an inhomogeneous environment such as an
interface. By testing β-lactoglobulin fibrils with varying average
thicknesses, we conclude that fibril thickness plays a determining
role in the propensity to form rings.
Free energy landscapes of proteins: insights from mechanical probes
Zu Thur Yew, Peter D Olmsted, and Emanuele Paci,
Advances in Chemical Physics: "Single-Molecule Biophysics: Experiment
and Theory" 146 (2012)
394-417.
Introduction; One-Dimensional Models of Mechanical Strength; Multidimensionality
of Energy Landscapes; Use of the Jarzynski Relation to Directly Determine Free
Energies; Conclusion.
Internal protein dynamics shifts the distance
to the mechanical transition state
DK West, E Paci, and PD Olmsted, Physical Review E 74 (2006)
061912.
Mechanical unfolding of polyproteins by force spectroscopy provides
valuable insight into their free energy landscapes. Most experiments
of the unfolding process have been fit to two-state and/or one
dimensional models, with the details of the protein and its dynamics
often subsumed into a zero-force unfolding rate and a distance x_u(1D)
to the transition state. We consider the entire phase space of a model
protein under a constant force, and show that x_u(1D) contains a
sizeable contribution from exploring the full multidimensional energy
landscape. This effect is greater for proteins with many degrees of
freedom that are affected by force; and surprisingly, we predict that
externally attached flexible linkers also contribute to the measured
unfolding characteristics.
Free energy of protein folding using the Jarzynski
equality
DK West, PD Olmsted, and E Paci,
Journal of Chemical Physics 125 (2006) 204910.
The equilibrium free energy difference between two long-lived
molecular species or "conformational states" of a protein (or any
other molecule) can in principle be estimated by measuring the work
needed to shuttle the system between them, independent of the
irreversibility of the process. This is the meaning of the Jarzynski
equality (JE), which we test in this paper by performing simulations
that unfold a protein by pulling two atoms apart. Pulling is performed
fast relative to the relaxation time of the molecule and is thus far
from equilibrium. Choosing a simple protein model for which we can
independently compute its equilibrium properties, we show that the
free energy can be exactly and effectively estimated from
nonequilibrium simulations. To do so, one must carefully and correctly
determine the ensemble of states that are pulled, which is more
important the farther from equilibrium one performs simulations; this
highlights a potential problem in using the JE to extract the free
energy from forced unfolding experiments. The results presented here
also demonstrate that the free energy difference between the native
and denatured states of a protein measured in solution is not always
equal to the free energy profile that can be estimated from forced
unfolding simulations (or experiments) using the JE.
Mechanical unfolding revisited through a simple
but realistic model
DK West, PD Olmsted, and E Paci,
Journal of Chemical Physics 124 (2006) 154909.
Single-molecule experiments and their application to probe the
mechanical resistance and related properties of proteins provide a new
dimension in our knowledge of these important and complex biological
molecules. Single-molecule techniques may not have yet overridden
solution experiments as a method of choice to characterize biophysical
and biological properties of proteins, but have stimulated a debate
and contributed considerably to bridge theory and experiment. Here we
demonstrate this latter contribution by illustrating the reach of some
theoretical findings using a solvable but nontrivial molecular model
whose properties are analogous to those of the corresponding
experimental systems. In particular, we show the relationship between
the thermodynamic and the mechanical properties of a protein. The
simulations presented here also illustrate how forced and spontaneous
unfolding occur through different pathways and that folding and
unfolding rates at equilibrium cannot in general be obtained from
forced unfolding experiments or simulations. We also study the
relationship between the energy surface and the mechanical resistance
of a protein and show how a simple analysis of the native state can
predict much of the mechanical properties of a protein.
Mechanical resistance of proteins explained using
simple molecular models
Daniel K. West, David J. Brockwell, Peter
D. Olmsted, Sheena E. Radford, and Emanuele Paci, Biophysical
Journal 90 (2006) 287-297.
Recent experiments have demonstrated that proteins unfold when two
atoms are mechanically pulled apart, and that this process is
different to when heated or when a chemical denaturant is added to the
solution. Experiments have also shown that the response of proteins to
external forces is very diverse, some of them being "hard", and
others "soft". Mechanical resistance originates from the presence of
barriers on the energy landscape; together, experiment and simulation
have demonstrated that unfolding occurs through alternative pathways
when different pairs of atoms undergo mechanical extension. Here we
use simulation to probe the mechanical resistance of six structurally
diverse proteins when pulled in different directions. For this, we use
two very different models: a detailed, transferable one, and a
coarse-grained, structure-based one. The coarse-grained model gives
results that are surprisingly similar to the detailed one and
qualitatively agree with experiment; i.e., the mechanical resistance
of different proteins or of a single protein pulled in different
directions can be predicted by simulation. The results demonstrate the
importance of pulling direction relative to the local topology in
determining mechanical stability, and rationalize the effect of the
location of importation/degradation tags on the rates of mitochondrial
import or protein degradation in vivo.
Mechanically unfolding the small, topologically simple protein L
David J Brockwell Godfrey S Beddard, Emanuele Paci, Dan K West, Peter
D Olmsted, D Alastair Smith, and Sheena E Radford,
Biophysical Journal 89 (2005) 506-513.
Beta-sheet proteins are generally more able to resist mechanical
deformation than a-helical proteins. Experiments measuring the
mechanical resistance of b-sheet proteins extended by their termini
led to the hypothesis that parallel, directly hydrogen-bonded terminal
b-strands provide the greatest mechanical strength. Here we test this
hypothesis by measuring the mechanical properties of protein L, a
domain with a topology predicted to be mechanically strong, but with
no known mechanical function. A pentamer of this small, topologically
simple protein is resistant to mechanical deformation over a wide
range of extension rates. Molecular dynamics simulations show the
energy landscape for protein L is highly restricted for mechanical
unfolding and that this protein unfolds by the shearing apart of two
structural units in a mechanism similar to that proposed for
ubiquitin, which belongs to the same structural class as protein L,
but unfolds at a significantly higher force. These data suggest that
the mechanism of mechanical unfolding is conserved in proteins within
the same fold family and demonstrate that although the topology and
presence of a hydrogen-bonded clamp are of central importance in
determining mechanical strength, hydrophobic interactions also play an
important role in modulating the mechanical resistance of these
similar proteins.
Pulling geometry defines the mechanical resistance of a
beta-sheet protein
DJ Brockwell, E Paci, RC Zinober, GS
Beddard, PD Olmsted, DA Smith, RN Perham, and SE Radford, Nature
Structural Biology 10 (2003) 731. Reviewed in Nature
Structural Biology 10 (2003) 674-676 (News and Views),
Science 301 (2003) 1291.
Proteins show diverse responses when placed under mechanical
stress. The molecular origins of their differing mechanical resistance
are still unclear, although the orientation of secondary structural
elements relative to the applied force vector is thought to have an
important function. Here, by using a method of protein immobilization
that allows force to be applied to the same all- protein, E2lip3, in
two different directions, we show that the energy landscape for
mechanical unfolding is markedly anisotropic. These results, in
combination with molecular dynamics (MD) simulations, reveal that the
unfolding pathway depends on the pulling geometry and is associated
with unfolding forces that differ by an order of magnitude. Thus, the
mechanical resistance of a protein is not dictated solely by amino
acid sequence, topology or unfolding rate constant, but depends
critically on the direction of the applied extension.
Unfolding Dynamics of Proteins Under Applied Force
DA
Smith, DJ Brockwell, RC Zinober, AW Blake, GS Beddard, PD Olmsted, and
SE Radford, Philosophical Transactions of the Royal Society A 361
(2003) 713-730. From the Royal Society Discussion Meeting, "Slow
Dynamics in Complex Systems", 25-26 September 2002.
Mechanically unfolding proteins: the effect of unfolding
history and the supramolecular scaffold
RC Zinober, DJ Brockwell,
GS Beddard, AW Blake, PD Olmsted, SE Radford, and DA Smith, Protein
Science 11 (2002) 2759-2765.
The mechanical resistance of a folded domain in a polyprotein of five
mutant I27 domains (C47S, C63S I27)5is shown to depend on the
unfolding history of the protein. This observation can be understood
on the basis of competition between two effects, that of the changing
number of domains attempting to unfold, and the progressive increase
in the compliance of the polyprotein as domains unfold. We present
Monte Carlo simulations that show the effect and experimental data
that verify these observations. The results are confirmed using an
analytical model based on transition state theory. The model and
simulations also predict that the mechanical resistance of a domain
depends on the stiffness of the surrounding scaffold that holds the
domain in vivo, and on the length of the unfolded domain. Together,
these additional factors that influence the mechanical resistance of
proteins have important consequences for our understanding of natural
proteins that have evolved to withstand force.
The Effect of Core Destabilisation on the Mechanical
Resistance of I27
DJ Brockwell, GS Beddard, J Clarkson, RC
Zinober, AW Blake, J Trinick, PD Olmsted, DA Smith, and SE Radford, Biophysical
Journal, 83 (2002) 458-472.
It is still unclear whether mechanical unfolding probes the same
pathways as chemical denaturation. To address this point, we have
constructed a concatamer of five mutant I27 domains (denoted (I27)5*)
and used it for mechanical unfolding studies. This protein consists of
four copies of the mutant C47S, C63S I27 and a single copy of C63S
I27. These mutations severely destabilize I27 (GUN = 8.7 and 17.9 kJ
mol1 for C63S I27 and C47S, C63S I27, respectively). Both mutations
maintain the hydrogen bond network between the A' and G strands
postulated to be the major region of mechanical resistance for
I27. Measuring the speed dependence of the force required to unfold
(I27)5* in triplicate using the atomic force microscope allowed a
reliable assessment of the intrinsic unfolding rate constant of the
protein to be obtained (2.0 × 103 s1). The rate constant of unfolding
measured by chemical denaturation is over fivefold faster (1.1 × 102
s1), suggesting that these techniques probe different unfolding
pathways. Also, by comparing the parameters obtained from the
mechanical unfolding of a wild-type I27 concatamer with that of
(I27)5*, we show that although the observed forces are considerably
lower, core destabilization has little effect on determining the
mechanical sensitivity of this domain.